Pathophysiologie des arteriellen O2-Status
Sauerstoffmangel im Gewebe: Gewebehypoxie
Im allgemeinen wird Hypoxie als Synonym für Sauerstoffmangel verwendet. Strenggenommen aber bezeichnet Hypoxie einen Abfall des pO2, in diesem Sinne spricht man z. B. von einer inspiratorischen, einer arteriellen oder einer Gewebehypoxie. Als Ursachen einer Gewebehypoxie kommen prinzipiell eine Störung der Durchblutung und eine Abnahme des arteriellen O2-Gehalts in Frage, in beiden Fällen wird das O2-Angebot reduziert. Grundsätzlich unterscheiden sich aber beide Ursachen insofern, als die Kompensationsmechanismen sehr unterschiedlich ausgeprägt sind.
Kompensationsmechanismen
Eine Abnahme der Durchblutung kann nur mit einer Zunahme der Utilisation kompensiert werden, die ihrerseits längerfristig über eine Verlagerung der O2-Bindungskurve optimiert werden kann. Längerfristig deshalb, weil die Zunahme der intraerythrozytären 2,3-DPG-Konzentration etwa 6 - 12 Stunden in Anspruch nimmt. Eine Verminderung des arteriellen O2-Gehalts hingegen kann mit einer Steigerung der Durchblutung, und zwar systemisch, Steigerung des HZV, oder lokal, Steigerung der Organdurchblutung, beantwortet werden. Zusätzlich kann auch hier eine Kompensation über eine Zunahme der Utilisation eingesetzt werden. Tab. Kompensation einer Gewebehypoxie gibt eine Übersicht hierzu.

Während die Utilisation je nach Organ, abgesehen von der Niere, nur um den Faktor 2 - 3 auf 100 % gesteigert werden kann, kann die Durchblutung unter physiologischen Bedingungen um den Faktor 4 - 5 gesteigert werden, dies gilt systemisch für das HZV und organspezifisch zumindest für das Myokard (Koronarreserve) und das ZNS. Es gibt keinen besseren Vasodilatator als eine Abnahme des lokalen Gefäß-pO2. Dies macht deutlich, warum eine Durchblutungsabnahme prinzipiell dramatischer verlaufen muss als z. B. eine akute oder erst recht eine chronische Anämie. Schließlich fällt auf, dass die Kompensation über eine Durchblutungssteigerung derjenigen einer Utilisationszunahme zeitlich immer vorgezogen wird, so als wollte der Organismus die "venöse Reserve", d. h. den nicht utilisierten venösen Sauerstoff, möglichst lange unangetastet lassen. Der Grund dafür dürfte sein, den venösen pO2 als treibende Kraft für die O2-Diffusion in das Gewebe möglichst hoch zu halten.
Differentialdiagnose der arteriellen Hypoxämie
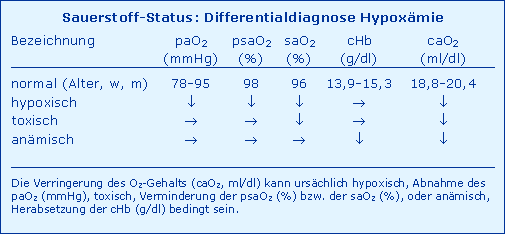
Die Nomenklatur des arteriellen O2-Status kennt prinzipiell vier pathophysiologische Charakteristika, nämlich eine
- Hypoxie als Abnahme des paO2
- Hypoxygenation als Verminderung der saO2 (oder der psaO2)
- Anämie als Herabsetzung der cHb
- Hypoxämie als Verringerung des caO2.
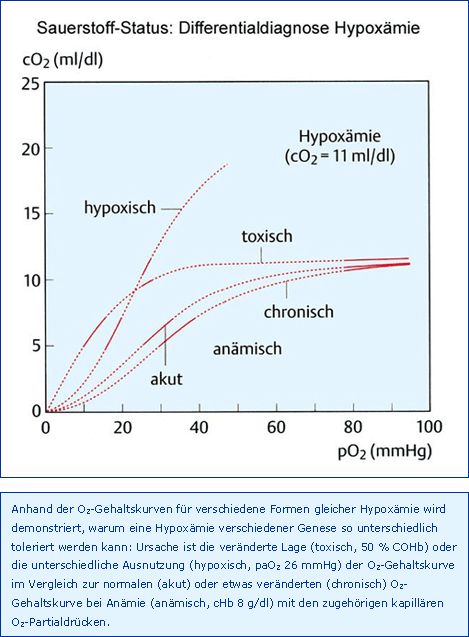
Die vier genannten Größen sind gemäß Abb. Determinanten wie eine Kausalkette aufzufassen, d. h. eine Hypoxie muss zu einer Hypoxygenation und Hypoxämie führen, eine Hypoxygenation kann hypoxisch oder toxisch sein und bedingt ebenfalls eine Hypoxämie und desgleichen die Anämie, die auch eine Hypoxämie verursacht. Somit kann die arterielle Hypoxämie bezüglich ihrer Ursache hypoxisch, toxisch oder anämisch sein, wie in Abb. Differentialdiagnose Hypoxämie dargestellt.
Diese Differenzierung ist keine akademische, sondern ist bezüglich ihrer diagnostischen und therapeutischen Konsequenzen zu berücksichtigen, wie aus Tab. Differentialdiagnose Hypoxämie eindeutig hervorgeht.
Hypoxische Hypoxämie
Die arterielle Hypoxie ist primär durch einen beim Patienten auftretenden Abfall des paO2 unter den Normalwertbereich von, je nach Alter, 78 - 95 mmHg verursacht und führt sekundär zu einer Abnahme der saO2 unter den Normalwert von 96 % bzw. tertiär des caO2 von 18,5 - 20,5 ml/dl je nach Geschlechtszugehörigkeit. Als Ursache kommt in Frage jede Störung der Lungenfunktion, der äußeren Atmung, des Ventilations-Perfusions-Verhältnisses oder auch der Beatmung. Das Ausmaß dieser hypoxischen Hypoxämie wird klinisch anhand der Hypoxygenation beurteilt: Gemäß der O2-Bindungskurve (Abb. O2-Bindungskurve) wird sich eine Hypoxie erst bei paO2-Werten unterhalb von 60 mmHg bemerkbar machen, also saO2-Werten unterhalb von 90 %. Da die saO2 selbst bei einem paO2 von 50 mmHg noch 85 % beträgt, können therapeutische Grenzwerte relativ großzügig angegeben werden. Der theoretisch mögliche Fall einer Hypoxygenation bei normalem paO2 infolge deutlicher Rechtsverlagerung der O2-Bindungskurve kann praktisch ausgeschlossen werden. Realistisch hingegen ist eine langfristige, über eine gesteigerte Erythropoese erfolgende Erhöhung der cHb mit der Folge, dass trotz bestehender Hypoxie und Hypoxygenation, paO2 und saO2 liegen unterhalb der Norm, die caO2 weitgehend normalisiert wird. Diese Polyglobulie findet sich daher bei chronischen Hypoxien und Hypoxygenationen, also nach längerfristigem Höhenaufenthalt, bei chronischen Lungenfunktionsstörungen oder persistierenden Herzfehlern mit Rechts-Links-Shunt.
Therapeutische Grenzwerte
Für den klinisch wichtigen Fall der akuten, arteriellen Hypoxie sollen therapeutische Grenzwerte entwickelt werden. Eine realistische Ableitung findet sich bei Nunn [1993]: Limitierendes Organ der akuten, arteriellen Hypoxie dürfte das Gehirn mit seiner speziellen O2-Versorgung sein, der Bewusstseinsverlust tritt bei einem pvO2 von 20 mmHg ein: Ohne Änderung der Durchblutung - unrealistisch - und ohne Bewusstseinsverlust wird ein paO2 von 36 mmHg (psaO2 68 %) abgeleitet, mit Verdoppelung der Durchblutung - realistisch - ein paO2 von 27 mmHg (psaO2 50 %). Eine pragmatische Herleitung wurde mit den pulsoxymetrischen psaO2-Werten von fast 500 Touristen unternommen, die sich in weniger als einer Stunde einer akuten Hypoxie als Flugpassagiere oder Höhentouristen unterwarfen [Zander, Mertzlufft 1996]: Die untersuchten Touristen im Alter von 10 - 90 Jahren tolerieren eine mittlere Abnahme der psaO2 auf ca. 75 % bzw. des paO2 auf 40 mmHg. Eine gegenüber der Normoxie durch die Hypoxie zusätzlich betonte Altersabhängigkeit der psaO2 kann nicht nachgewiesen werden.
Therapeutische Grenzwerte für eine akute, arterielle Hypoxie, nämlich psaO2 90 % bzw. paO2 60 mmHg (fakultativ) und 75 % bzw. 40 mmHg (obligatorisch), können als gut begründet angesehen werden und gelten für Patienten beiderlei Geschlechts mit weitgehend normaler Hb-Konzentration in körperlicher Ruhe. Sie können dem Anästhesisten ein eventuelles (fakultatives) oder unbedingtes (obligatorisches) Handeln nahe legen, z. B. im Sinne einer Erhöhung der inspiratorischen O2-Konzentration. Bei Annahme dieses limitierenden paO2 von 40 mmHg (psaO2 75 %) sind für den klinischen Alltag weitere Sicherheitsmarchen vorhanden, die zusätzliche Unwägbarkeiten für das Gehirn mit Sicherheit kompensieren können: Eine leichte Anämie (Abnahme der cHb auf 10 - 12 g/dl mit Zunahme der avDO2), eine Zerebralsklerose (verminderte hypoxische Zunahme der Durchblutung) oder eine Hypokapnie (Linksverlagerung der O2-Bindungskurve, hypokapnische Durchblutungsabnahme).
Toxische Hypoxämie
Hypoxygenationen mit normalem paO2 deuten immer auf eine toxische Hypoxämie hin, d. h. das O2-Bindungsvermögen des Hämoglobins ist durch das Auftreten von Dyshämoglobinen reversibel eingeschränkt. Als Ursachen kommen in Frage eine Kohlenmonoxid-Belastung (CO) mit der Bildung von Carboxy-Hämoglobin (COHb) oder die Oxidation des Häm-Eisens mit der Umwandlung von Hämoglobin zu Hämiglobin (MetHb). Beide Störungen sind reversibel, allerdings mit unterschiedlicher Kinetik: In körperlicher Ruhe wird CO mit einer Halbwertszeit von ca. 8 h abgeatmet; MetHb wird durch die Wirkung intraerythrozytärer Reduktionssysteme (MetHb-Reduktase, Glutathion) mit einer Halbwertszeit von ca. 2 h zu Hämoglobin reduziert.
Kohlenmonoxid-Intoxikation
Im Rahmen einer CO-Intoxikation (Rauchvergiftung, Autoabgase von Motoren ohne Katalysator) oder einer chronischen CO-Exposition, wie sie beim Tabakrauchen zu beobachten ist, wird ein unterschiedlicher Anteil des Hämoglobins mit CO besetzt. Die am Abend bei Zigarettenrauchern gemessenen COHb-Konzentrationen liegen zwischen 17 % [Zander, Mertzlufft 1991] und 22 % [Pankow 1981] als Maximalwerte. Die im Zigaretteninhalat gemessenen CO-Konzentrationen liegen mit 4 - 6 % höher als der maximal zulässige Wert für Autoabgase von 3 %.
Die Problematik von CO besteht in seiner sehr hohen Affinität zu Hb, die im Vergleich zu O2 im relevanten Bereich (5 % COHb bzw. O2Hb) etwa 350 mal größer ist. Die Ursache dafür ist die hyperbelförmige COHb-Bindungskurve, bereits 0,02 mmHg pCO (entsprechend der maximal zulässigen Arbeitsplatzkonzentration von 0,003 % CO) erzeugen bei mehrstündigem Aufenthalt 5 % COHb im Blut.
Die Wirkung von CO ist zweifacher Natur: Einmal wird Hb partiell für den O2-Transport ausgeschaltet, zum anderen - dies macht die eigentliche Wirkung aus - wird die Affinität des noch verbleibenden, freien Hb deutlich erhöht, weil das freie Hb mit zunehmendem COHb eine hyperbelförmige Bindungskurve annimmt.
Besonders problematisch wird COHb dann, wenn es unter der Schwangerschaft im mütterlichen Kreislauf erscheint, weil vor allem die hohe Hb-Konzentration des Feten dafür sorgt, dass CO praktisch vollständig plazentar auf die fetale Blutseite transportiert wird.
Soll im Rahmen einer Therapie die Halbwertszeit für CO vermindert werden, so kann dies durch Gabe von reinem O2 erfolgen: Der sogenannte Halbsättigungsdruck der CO-Bindungskurve, d. h. der für die Einstellung von 50 % COHb notwendige pCO, wird von nur 0,35 auf immerhin 1,8 mmHg erhöht, wenn der paO2 therapeutisch von 90 auf 500 mmHg erhöht werden kann. So wird durch Gabe von reinem O2, d. h. durch normobare Hyperoxie, das toxische CO aus seiner Bindung zu Hb verdrängt, eine hyperbare O2-Therapie in einer Überdruckkammer kann diesen Prozess noch erheblich verstärken.
Intoxikation mit Met-Hämoglobin-Bildnern
Eine Erhöhung der MetHb-Konzentration wird immer dann auftreten, wenn oxidierende Substanzen die Möglichkeit bekommen, Hämoglobin (Fe2+) in Hämiglobin (Fe3+) bzw. MetHb umzuwandeln. Die derzeit interessantesten Substanzen dürften bestimmte Lokalanästhetika einerseits und Nitrat aus dem Trinkwasser andererseits sein, das nach Darmpassage als das eigentliche Oxidationsmittel Nitrit im Blut erscheinen kann. Die sogenannte "Brunnenwasser-Blausucht" von Säuglingen und Kleinkindern ist ein Beispiel. Das Lokalanästhetikum Prilocain erzeugt über seine Abbau-Metabolite schon in therapeutischen Dosen bis 600 mg MetHb bis zu einer Konzentration von über 20 % [Biscoping, Hempelmann 1985; Geiger et al. 1989; Thiessen et al. 1984], der höchste veröffentlichte Wert wird mit 35,7 % bei einem Säugling angegeben [Biscoping, Hempelmann 1985]. Da die entstehende Zyanose klinisch meist unerkannt bleibt, die MetHb-Konzentration nur langsam über mehrere Stunden ansteigt und eine Diagnostik von MetHb nur in den seltensten Fällen möglich ist, wird diese toxische Hypoxämie in der anästhesiologischen Praxis meist verharmlost oder übersehen.
Zwei Patientengruppen soll hierbei besonderes Augenmerk geschenkt werden: Eine MetHb-Bildung bei Anämie-Patienten führt zu einer Kombination aus anämischer plus toxischer Hypoxämie, die eine fatale Gewebehypoxie erzeugen kann. Da bei Säuglingen und Kleinkindern die intraerythrozytär vorhandenen Reduktionssysteme schlechter ausgebildet sind als beim Erwachsenen, verdoppelt sich die Halbwertszeit in etwa von ca. 2 auf 4 h mit der Folge, dass die MetHb-Rückbildung deutlich verzögert ist.
Neben den früher häufig bei Methämoglobinämie eingesetzten Therapeutika Methylenblau und Ascorbinsäure wird heute Toluidinblau bevorzugt. Hinzuweisen ist allerdings auf die Tatsache, dass die Gabe von Toluidinblau als einer intensiv gefärbten Substanz jede Art von Photometrie für Minuten bis Stunden stört, insbesondere die für den Nachweis von MetHb heute zu empfehlenden Häm-Oxymeter.
Therapeutische Grenzwerte
Die Angabe von therapeutischen Grenzwerten ist bei der toxischen Hypoxämie aus drei Gründen problematisch: Einmal liegt in den seltensten Fällen ein Messwert für COHb oder MetHb beim Patienten vor, und die üblichen Methoden Blutgasanalyse (paO2) und Pulsoxymetrie (psaO2) täuschen mit normalen, d. h. normoxischen Werten, über die möglicherweise bedrohliche Hypoxämie des Patienten hinweg. Zum anderen deutet das klinische Bild der "kirschroten" Haut des COHb-Patienten und der "schokoladenbraunen" Zyanose des MetHb-Patienten nicht gerade auf einen O2-Mangel hin.
Schließlich bestehen zumindest bei der Prilocain-induzierten MetHb-Intoxikation - vielleicht deshalb - unter Anästhesisten große Meinungsunterschiede bezüglich der Notwendigkeit einer therapeutischen Intervention: Einmal bedarf eine MetHb-Konzentration von mehr als 20 % einer Behandlung [Thiessen et al. 1984], ein anderes Mal wird die Notwendigkeit einer Therapie selbst bei 35,7 % MetHb bei einem Säugling verneint [Biscoping, Hempelmann 1985].
Tatsache ist, wie in Abb. Differentialdiagnose Hypoxämie dargestellt, dass die Problematik der toxischen Hypoxämie in der Linksverlagerung der O2-Gehaltskurve des noch funktionstüchtigen Hämoglobins besteht und deshalb eine COHb- bzw. MetHb-Konzentration von etwa 60 %, d. h. 40 % freies Hb, als letal angesehen werden muss [Pankow 1981]. Zum Vergleich: Ein Patient mit einer chronisch-anämischen cHb von 6 g/dl, also auch nur 40 % des Normalwertes, übt seinen Beruf aus, wie sehr viele Dialysepatienten vor der Einführung von Erythropoetin demonstriert haben. Da bereits bei COHb- oder MetHb-Konzentrationen von mehr als 15 % uncharakteristische Symptome wie Kopfschmerzen, Schwäche, Müdigkeit und Schwindel auftreten, sollten 10 % Dyshämoglobine als fakultativer Grenzwert aufgefasst werden.
Als obligatorischer Grenzwert kann ein Wert von 20 % Dyshämoglobin empfohlen werden, er muss wegen der Linksverlagerung der O2-Gehaltskurve sensibler ausfallen als der einer hypoxischen Hypoxämie mit 25 % Verlust (saO2 75 %).
Anämische Hypoxämie
Die anämische Hypoxämie, Abnahme des caO2 als Folge einer Verminderung der cHb, ist gekennzeichnet durch einen normalen paO2 und damit saO2 (Normoxie) und ist bezüglich der kapillären O2-Utilisation ein besonders günstiger Sonderfall, wie in Abb. Differentialdiagnose Hypoxämie dargestellt. Da die anämische Hypoxämie die häufigste klinische Störung des arteriellen O2-Status ist, muss sie ausführlicher als die Hypoxie und die Toxämie besprochen werden, allerdings mit der Beschränkung auf die normovolämische, normoxische, anämische Hypoxämie, oder kurz Anämie bzw. Hämodilution.
Ziel jeder Kompensation einer Anämie muss es sein, den O2-Verbrauch des Organismus und seiner Organe sicherzustellen bzw. konstant zu halten. Dazu kann die hämodynamische Kompensation über eine Steigerung des Schlagvolumens oder eine Frequenzzunahme des Herzens erfolgen, die utilisatorische Kompensation nimmt in Kauf, dass die gemischtvenöse O2-Konzentration mehr oder weniger deutlich abfällt.
Bemerkenswert dabei ist, dass die genannten Mechanismen insofern in einer bestimmten Reihenfolge ablaufen, als primär eine Zunahme des Schlagvolumens beobachtet wird, erst sekundär eine Frequenzsteigerung und in Verbindung mit dieser hämodynamischen Kompensation tertiär eine vermehrte Utilisation des venösen bzw. kapillären Blutes. Schließlich kann unter "chronischen" Bedingungen, wenn die Abnahme der cHb schon seit 6 - 12 Stunden besteht, eine quartäre Kompensation insofern erfolgen, als über eine Vermehrung der 2,3-DPG-Konzentration des Blutes eine Rechtsverlagerung der O2-Bindungskurve eingestellt wird, mit dem Ziel, die O2-Abgabe vom Blut an das Gewebe über eine Erhöhung des kapillären pO2 zu verbessern (s. Abb. Transportprozesse).
Für die tägliche klinische Praxis ist es zum Beispiel von besonderer Bedeutung, dass bis zu einer cHb von ca. 7,5 g/dl die hämodynamische Kompensation bei konstanter Herzfrequenz allein über eine Zunahme des Schlagvolumens erfolgt. Bisweilen sind Extrembetrachtungen sehr hilfreich, so auch hier die Situation einer extremen Anämie, wie sie bei Verweigerung der Transfusion beobachtet werden kann, nämlich Hb-Konzentrationen von nur noch 1,5 g/dl, die teilweise überlebt wurden [Zander 1996 (B)]. Tatsächlich kann man im Tierversuch nachweisen [8 Tierversuchsreihen in Zander 1996 (B)], dass unter isovolämischer Hämodilution die Gesamt-O2-Aufnahme der Tiere mit einer Streuung von ± 10 % bis zu einer cHb von ca. 3 g/dl konstant bleibt. Erst bei dieser cHb unterschreitet der gemischtvenöse pO2 die möglicherweise kritische Grenze von 25 - 35 mmHg. Auch diese Befunde belegen, dass die anämische Hypoxämie erstaunlich gut toleriert wird, deutlich besser als andere Formen der Hypoxämie, insbesondere wegen der praktisch 100-%igen Utilisation des angebotenen O2 in den Kapillaren fast aller Organe. Bei einem pvO2 von 25 mmHg, einer svO2 von ca. 50 % und einer cHb von 3 g/dl beträgt der cvO2 noch ca. 2 m/dl, also nur noch 10 % des arteriellen Normalwertes.
Wenn trotz dieser Befunde für Computersimulationen einer Hämodilution ein kritischer gemischtvenöser pO2 von 35 mmHg (Lundsgaard-Hansen et al. 1989] oder eine gemischtvenöse sO2 von mindestens 70 % [Hoeft et al. 1995] als limitierender Faktor eingesetzt wird, kann dem nicht gefolgt werden: Die Annahme, eine kapilläre O2-Utilisation sei unterhalb eines venösen pO2 von 35 mmHg oder einer sO2 von 70 % nicht mehr möglich - die ungenutzte venöse O2-Reserve fließt am hypoxischen Gewebe vorbei - ist sicher nicht haltbar.
Das Herz als limitierendes Organ
Limitierendes Organ für jede Anämie dürfte mit Sicherheit das Myokard sein, weil es unter physiologischen Bedingungen die größte avDO2 aufweist, es utilisiert bereits in körperlicher Ruhe 60 % des arteriell angebotenen Sauerstoffs (s. Tab. O2-Utilisation).
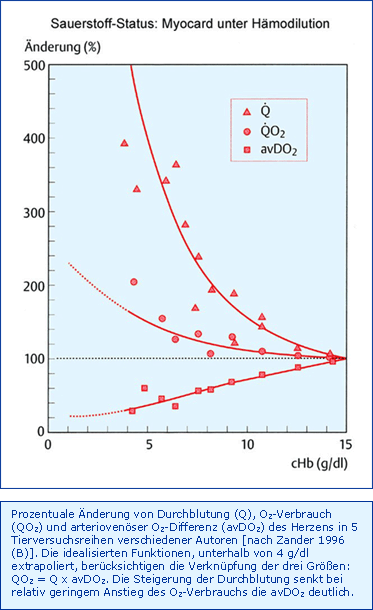
Die Antwort des Myokards auf eine isovolämische Hämodilution ist in Abb. Myokard unter Hämodilution wiedergegeben. Eine Hämodilution bis zu einer cHb von 7,5 g/dl führt zu einer Steigerung der Koronardurchblutung, wegen der Viskositätssenkung weitgehend ohne Vasodilatation, um nur ca. 100 %, während die physiologische Koronarreserve maximal 500 % beträgt. Entscheidend ist die Frage, in welchem Ausmaß das Myokard seinen eigenen O2-Verbrauch im Dienste des Gesamtorganismus (Steigerung des HZV) steigern muss: Bei der gleichen Hämodilution auf 7,5 g/dl kommt es zu einer Zunahme des HZV um 50 - 70 % aber einer O2-Verbrauchszunahme von nur 0 bis 37 %. Der vermeintliche "Nachweis" einer Abnahme des myokardialen O2-Verbrauchs um ca. 10 % unter vergleichbaren Bedingungen musste angezweifelt werden [Zander 1997 (A)]. Somit bleibt festzuhalten, dass das intakte Myokard als limitierendes Organ ohne Schwierigkeiten eine Senkung der cHb auf ca. 7,5 g/dl durch Steigerung des HZV (Viskositätserniedrigung gleich Nachlastsenkung) kompensieren kann, ohne die Koronardurchblutung unphysiologisch steigern zu müssen. Tatsache aber ist auch, dass jede weitere Hämodilution nur noch von der weiteren fraglichen Steigerung der Koronardurchblutung abhängen muss.
Gibt es ein Optimum für die Hb-Konzentration?
Die Frage nach einem möglichen Optimum für die Hb-Konzentration eines normovolämischen, normoxischen Patienten in körperlicher Ruhe kann nicht identisch sein mit der Frage der therapeutischen Grenzwerte der cHb, d. h. unterhalb welcher cHb eine Transfusion von Erythrozyten zu empfehlen ist.
Einerseits stellt die physiologische cHb, nämlich 13,9 g/dl bei der Frau und 15,3 g/dl beim Mann, einen evolutionären Kompromiss für verschiedenste Situationen dar: Für sehr unterschiedliche körperliche Aktivitäten (Ruhe, maximale Arbeit), Umgebungsbedingungen (Fetalzeit, Höhenaufenthalt), Hormonstatus (Alter, Geschlecht, Schwangerschaft) und Störgrößen (z. B. Rauchen) wurde eine gemeinsame cHb vom Organismus entwickelt. Dieser Kompromiss wäre als eine Art von Optimum aufzufassen, muss deshalb aber nicht zwangsläufig als optimale cHb für einen Patienten gelten. Andererseits muss eine Änderung der cHb aus folgenden Gründen zwangsläufig ein Optimum bedingen: Einmal bestimmt sie wesentlich die caO2 und zum anderen beeinflusst sie sehr deutlich die Viskosität des Blutes.
Eine Abnahme der cHb zum Beispiel wird deshalb zwei Effekte haben, nämlich eine Abnahme des O2-Angebotes, weil die caO2 abnimmt, und eine Zunahme des O2-Angebotes, weil die Viskositätsabnahme des Blutes, d. h. Verminderung des peripheren Widerstandes, automatisch zu einer Zunahme des HZV führt. Diese beiden Effekte bedingen somit, dass bei Abnahme der cHb mit einem Optimum für das O2-Angebot zu rechnen sein muss.
Mit den klassischen Untersuchungen über die akute isovolämische Hämodilution im Tierexperiment wurde versucht [Messmer et al. 1972; Sunder-Plassmann et al. 1971], dieses Optimum zu belegen: Das systemische Sauerstoffangebot als Produkt von HZV und caO2 sollte beim Hämatokrit von ca. 30 % bzw. einer cHb von ca. 10 g/dl ein Maximum von etwa 110 % in Relation zum Ausgangswert aufweisen. Das HZV sollte somit kompensatorisch überproportional gesteigert werden, was auf die deutliche Senkung der Blutviskosität zurückgeführt wurde. Die Analyse von Literaturdaten hat jedoch gezeigt, dass lediglich eine Arbeitsgruppe Befunde für diese These vorgelegt hat, allerdings nur Daten, die in den genannten Publikationen nicht belegt sind und einer wissenschaftlichen Nachprüfung nicht standhalten [Zander 1999].
Trotzdem gilt, dass die hämodynamische Kompensation einer Abnahme der normalen cHb bzw. caO2 in einem weiten Bereich fast vollständig durch eine Steigerung des HZV kompensiert wird. Dies gilt auch für jedes einzelne Organ mit seiner Durchblutung. Andere Optima für die cHb wurden aber in folgenden Situationen nachgewiesen [Zander 1998 (A)]:
- Bei der Therapie der renalen Anämie mit Hilfe von Erythropoetin hat sich eingebürgert, eine cHb von 10 - 11 g/dl anzustreben (Zielhämatokrit 30 - 35 %), da oberhalb dieses Wertes keine weiteren Leistungssteigerungen mehr zu erzielen sind sondern eher nachteilige hämodynamische Auswirkungen beobachtet werden.
- Nach Auswertung von ca. 50.000 Geburtsprotokollen weißer und schwarzer Frauen ließ sich nachweisen, dass bezüglich des Geburtsergebnisses (lebend, termingerecht, Mindestgewicht 2.500 g, Apgarwert über 3 usw.) ein eindeutiges Optimum für die cHb von 11 g/dl (Schwarze) und 12 g/dl (Weiße) nachzuweisen ist.
- Für die tägliche Praxis einer orthopädischen Klinik wird eine optimale cHb von 10 g/dl ("optimaler Hkt 30%") empfohlen und angestrebt.
Bleibt die Frage nach der Möglichkeit, dieses Optimum zum klinischen Richtwert zu erheben und, in welcher Form auch immer, zu bestätigen oder zu widerlegen. Zu diesem Zweck wurden solche Untersuchungen am Menschen zusammengefasst, die im Rahmen einer Hämodilution oder einer Hämokonzentration Belege für eventuelle Vor- oder Nachteile einer Änderung der cHb erbringen sollten [Zander 1998 (A)]. Es ist offensichtlich, dass solche Belege weder für eine Hämokonzentration noch für eine Hämodilution erbracht wurden: Eine deutliche Steigerung des O2-Angebotes ist zwar für die Hämokonzentration nachweisbar, die O2-Aufnahme oder der -Verbrauch, die Herzfrequenz, die Laktatkonzentration oder der Base Excess änderten sich aber nicht. Das gleiche gilt auch für die Hämodilution: Mit einer Ausnahme änderte sich in keinem Falle der O2-Verbrauch signifikant, der Herzindex nahm erwartungsgemäß zu, es kam zu keiner Änderung der Laktatkonzentration (bei Hämodilution bis auf 5,9 oder 3,0 g/dl), in einem Falle nur zu der sogenannten, nicht hypoxisch bedingten Verdünnungsazidose.
Auch eine sehr breit angelegte Multicenter-Studie an 25 Intensivstationen, bei der nach strengen Kriterien von ursprünglich ca. 6.500 Patienten noch 848 in die Auswertung einbezogen wurden [Hébert et al. 1999], kam für kritisch Kranke zu dem Ergebnis, dass eine restriktive Transfusionsstrategie (418 Patienten: cHb = 8,5 ± 0,7 g/dl) im Vergleich zu einer liberalen Strategie (420 Patienten: cHb = 10,7 ± 0,7 g/dl) genauso effektiv und sogar teilweise überlegen war. Nicht nur die Zahl der transfundierten Erythrozytenkonzentrate pro Patient konnte von 5,6 ± 5,3 auf 2,6 ± 4,1 gesenkt werden (33 % der Patienten der restriktiven Gruppe erhielten überhaupt kein EK), sondern auch die Mortalität über 30 und 60 Tage war bei gleichen Liegezeiten geringer, ebenso die Komplikationsrate, auch der Myokardinfarkte während des Krankenhausaufenthaltes. Eine mögliche Ausnahme gilt nach den Autoren lediglich für Patienten mit akutem Myokardinfarkt und Angina pectoris.
Somit gilt bis zum Beweis des Gegenteils: Im Bereich einer cHb von 7 - 15 g/dl profitiert kein Patient von einer Transfusion und erfährt keine Nachteile durch eine unterlassene Transfusion, vielleicht mit Ausnahme des Patienten mit koronarer Herzkrankheit.
Therapeutische Grenzwerte
Es gibt wohl keinen Bereich der Medizin, in welchem so viele therapeutische Grenzwerte veröffentlicht worden sind wie für die Anämie oder Hämodilution. Ziel derartiger Grenzwerte soll sein, dem klinisch tätigen Arzt eine Orientierung anzubieten, die trotzdem eine möglichst große Sicherheit gewährleistet. Neben einer in Ausnahmefällen möglichen cHb von 3,0 g/dl [Zander 1996 (B)] und einer als klinisch tolerabel bezeichneten von 6,5 g/dl [Mehrkens et al. 1990] existiert die Empfehlung des amerikanischen NIH aus dem Jahre 1989 [NIH 1989], oberhalb einer cHb von 7,0 g/dl kein Blut oder Erythrozyten zu transfundieren, es sei denn, Symptome eines O2-Mangels seien nachweisbar (EKG-Kontrolle, Zunahme der Herzfrequenz, des negativen Base Excess oder der Laktat-Konzentration).
Für die tägliche klinische Praxis kann ein gut begründeter Richtwert einer optimalen cHb von 10 g/dl empfohlen werden, der gleichermaßen für Hämodilution und Hämokonzentration gelten kann. Es gibt keinen Anlass, für den geriatrischen Patienten Ausnahmen von diesen Werten zu empfehlen [Zander 1998 (B)]. Eine Transfusion von Blut oder Erythrozyten im Bereich der cHb von 7 - 10 g/dl macht im Sinne einer forensischen Rechtfertigung den Nachweis von Hypoxie-Zeichen zur Transfusionsindikation erforderlich, was unterhalb von 7,0 g/dl entfällt.
Synopsis der therapeutischen Grenzwerte der arteriellen Hypoxämie
Anhand der im einzelnen begründeten therapeutischen Grenzwerte der arteriellen Hypoxämie, zusammengefasst in Tab. Synopsis Grenzwerte Hypoxämie, werden die Unterschiede zwischen den verschiedenen Formen deutlich, wie dies auch schon in Abb. Differentialdiagnose Hypoxämie dargestellt wurde.

Wegen der sehr unterschiedlichen kapillären O2-Partialdrücke muss sich eine arterielle Hypoxämie je nach Ursache unterschiedlich auswirken: Die größte Toleranz besteht gegenüber der anämischen Hypoxämie mit einer 50%igen Reduzierung des caO2, die geringste gegenüber der toxischen Hypoxämie mit einer nur 20%igen Abnahme des caO2. Fatal dabei ist, dass nur die hypoxische Hypoxämie klinisch durch eine Abnahme der pulsoxymetrisch gemessenen psaO2 auffällt.
Klinik einer Verlagerung der O2-Bindungskurve
Es ist denkbar, dass eine Linksverlagerung der O2-Bindungskurve und damit der O2-Gehaltskurve zu einer Verschlechterung der Gewebeversorgung führt und umgekehrt eine Rechtsverlagerung zu einer Beeinträchtigung der alveolären O2-Aufnahme. Unter physiologischen Bedingungen kommt beides nicht vor: Eine durch Hyperventilation bedingte Höhenalkalose z. B. wird in einigen Stunden über eine Zunahme der 2,3-DPG-Konzentration weitgehend aufgehoben, die bei allen Anämie-Formen zu beobachtende Rechtsverlagerung der O2-Gehaltskurve erfolgt nur soweit, dass beim physiologischen paO2 von ca. 90 mmHg keine Abnahme der saO2 in Kauf genommen werden muss. Da solche Verlagerungen bisweilen durch ärztliche Maßnahmen erfolgen, soll darauf eingegangen werden, die Verlagerungen durch Dyshämoglobine wurden bereits besprochen.
Hypothermie
Jede Hypothermie führt automatisch zu einer Linksverlagerung der O2-Bindungskurve, eine Gefahr für den Patienten entsteht dadurch aber nicht. Da die Hypothermie, als therapeutisches Ziel, den O2-Verbrauch des Patienten und aller Organe deutlich einschränkt, nach der RGT-Regel (Reaktions-Geschwindigkeits-Temperatur-Regel) nimmt der O2-Verbrauch bei Abkühlung um 10 °C auf ca. 30 % des Ausgangswertes ab, spielt die verschlechterte O2-Abgabe vom hypothermen Blut nur eine nachgeordnete Rolle. Die verbesserte O2-Aufnahme in der Lunge ist beim beatmeten Patienten uninteressant.
Gelagertes Blut
Gelagertes Vollblut oder Erythrozytenkonzentrate weisen aufgrund des unphysiologisch sauren Milieus der Stabilisatorlösungen bereits direkt nach der Abnahme beim Spender einen pH-Wert zwischen 6,6 und 6,8 auf. Diese Azidose führt innerhalb weniger Tage Lagerung zu einem drastischen 2,3-DPG-Verlust der Erythrozyten mit der Folge einer deutlichen Linksverlagerung der O2-Bindungskurve, die dann nach Transfusion beim Patienten zum Tragen kommt: Nachdem die Erythrozyten im Patienten sofort bezüglich ihrer Azidose normalisiert werden, bleibt die Linksverlagerung der O2-Bindungskurve noch für Stunden erhalten, so lange nämlich, bis die 2,3-DPG-Konzentration wieder normalisiert worden ist. Stammt das Blut von einem Raucher mit z.B. 10 % COHb, wird dieser Effekt der Linksverlagerung noch verstärkt, d. h. der Halbsättigungsdruck fällt von 26 auf 18 mmHg (2,3-DPG-Verlust) und zusätzlich weiter auf 14 mmHg (10 % COHb) ab [Zander 1988]. Dieser Halbsättigungsdruck entspricht in etwa dem einer Blutprobe mit 50 % COHb, was beim Patienten mit CO-Intoxikation in etwa letal wäre. Für einige Indikationen wird daher immer Frischblut eingesetzt werden müssen.
Permissive Hyperkapnie
Bei der sogenannten permissiven Hyperkapnie wird im Rahmen der künstlichen Beatmung eine deutliche Erhöhung des paCO2 in Kauf genommen, nachdem gezeigt werden konnte, dass diese Hyperkapnie ohne Probleme toleriert werden kann. Natürlich führt die Hyperkapnie zu einer Rechtsverlagerung der O2-Bindungskurve mit der Gefahr der Hypoxygenation, also der Abnahme der saO2.
Da aus der sehr zahlreichen Literatur über die apnoische Oxygenation am Menschen bekannt ist, dass der Mensch unter hyperoxischer Hyperkapnie CO2-Partialdrücke bis zu 300 mmHg überlebt, solange der paO2 wenigstens 300 mmHg beträgt, können die für eine saO2 von 95 % notwendigen paO2-Werte wie folgt benannt werden [Zander, Mertzlufft 1994 (B): Von paO2 75 mmHg bei paCO2 40 mmHg bis zu paO2 105 mmHg bei paCO2 130 mmHg, also insgesamt Werte, die durch Erhöhung der FIO2 leicht einzustellen sind.
Klinik einer therapeutischen Hyperoxie
Mit einer Hyperoxie wird versucht, die O2-Versorgung des möglicherweise mangelversorgten Gewebes zu normalisieren oder zu verbessern. Prinzipiell kann dabei normobar, also bei normalen Druckverhältnissen, oder hyperbar vorgegangen werden, d. h. O2-Applikation in einer Überdruckkammer. Aus naheliegenden Gründen, eine Überdruckkammer steht nur in seltenen Fällen zur Verfügung, soll nur die normobare Hyperoxie besprochen werden.
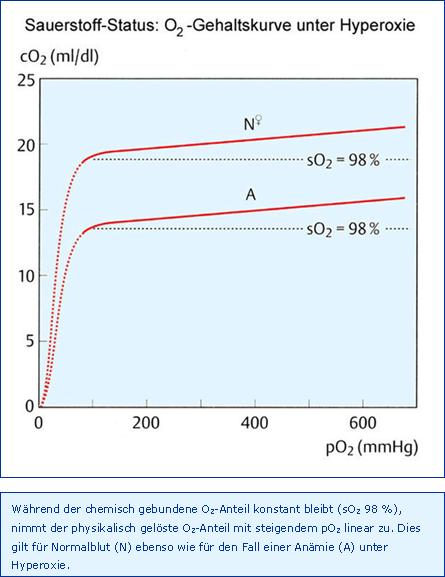
Der maximal erreichbare paO2 kann nach Auswaschen von N2 aus Lunge (ca. 1 min) und Gewebe (ca. 15 min) theoretisch 673 mmHg betragen, nämlich paO2 = pAO2 = pB 760 mmHg - pACO2 40 mmHg - pAH2O 47 mmHg, wenn der Barometerdruck pB 760 mmHg und die AaDO2 0 mmHg betragen. Somit erhöht sich der paO2 ganz erheblich mit der Folge, dass sich über den chemisch an Hb gebundenen O2 hinaus jetzt nur der physikalisch gelöste Anteil im Blut linear erhöht, wie dies in Abb. O2-Gehaltskurve unter Hyperoxie dargestellt ist. Auf diese Weise können immerhin bei einem paO2 von 625 mmHg 2,3 ml/dl O2 im Blut gelöst werden (O2-Löslichkeit 0,0037 ml/dl/mmHg bei cHb 15 g/dl), im Vergleich zu 0,3 ml/dl beim physiologischen paO2 von 90 mmHg, immerhin ein Zuwachs von 2 ml/dl. Unter hyperbaren Bedingungen von 2 bar (Gesamtdruck 3 bar) bedeutet dies, dass ein Patient mit einem caO2 von 6 ml/dl ohne Hb überlebt, was z. B. für die Therapie einer CO-Intoxikation lebensrettend sein kann.
Unter normobaren, klinischen Bedingungen kann über die Hyperoxie bei einer akuten Anämie kurzfristig ein Hb-Defizit von 1,5 g/dl ersetzt werden, da 1,5 g Hb gerade 2 ml/dl O2 transportieren können. Zusätzlich zur Erhöhung der caO2 bewirkt eine therapeutische Hyperoxie natürlich auch eine deutliche Steigerung des paO2 mit seinerseits Folgen für die O2-Versorgung.
Während in allen Organen unter Hyperoxie eine Durchblutungsabnahme um 10 - 15 % zu beobachten ist [Zander 1981], die caO2 nimmt dabei um ca. 10 % zu, wird für die Lunge der umgekehrte Effekt einer Vasodilatation beschrieben, der auch therapeutisch vielfältig genutzt wird [Pitton et al. 1998]. Wegen der Gefahr einer O2-Intoxikation gilt es als akzeptiert, dass eine Gabe von 100 % O2 nur maximal 24 h und eine von 50 % O2 nur maximal 48 h angewandt werden sollte.